







COVID-19: descifrando el origen

El origen del coronavirus del síndrome respiratorio agudo severo 2 (SARS-CoV-2), el agente de la enfermedad por coronavirus 2019 (COVID-19), es controvertido. La pandemia de la COVID-19 no solo tiene efectos dramáticos sobre las personas, sino que también produce efectos irreversibles en la sociedad, por ejemplo, en la economía, en la política, en las relaciones personales. De repente, el coronavirus llegó y nadie sabe de dónde. Lo que es verdad que ha llegado para quedarse. Establecer el origen del SARS-CoV-2 es un reto, el cual está ligado al origen de otros coronavirus relacionados. En el presente trabajo se ha abordado este reto mediante una aproximación bioinformática. El objetivo es intentar encajar las piezas del rompecabezas. La fuente de información ha sido las bases de datos del Centro Nacional de Información Biotecnológica (NCBI) (https://www.ncbi.nlm.nih.gov/) y la metodología se ha basado en los recursos bioinformáticos del propio NCBI y del Laboratorio Europeo de Biología Molecular ( EMBL) (https://www.embl.de/). En el espíritu del trabajo, ha estado siempre la reproducibilidad de los resultados, y mantener un debate sobre el origen del SARS-CoV-2.
¿Qué es lo aportado de nuevo con respecto al conocimiento previo?
Los resultados revelan la presencia de unos marcadores genéticos (fingerprints) en las secuencias de los genomas de los coronavirus relacionados con la COVID-19 y establecen una relaciones filogenéticas, que no se han sido descritas hasta la fecha:
• Primero, utilizando la herramienta de búsqueda de alineación local básica (Basic Local Alignment Search Tool, BLAST) (https://blast.ncbi.nlm.nih.gov/Blast.cgi) del NCBI, se identificaron claramente tres marcadores genéticos en las secuencias de los genomas de coronavirus relacionados con la COVID-19 (Tabla 1): (i) al inicio del genoma, también en el inicio del gen Orf1ab de la replicasa de ARN, donde se codifica el Macrodominio N-terminal (módulo de unión de ATP); (ii) en el gen S, donde se codifica el dominio N-terminal y en el dominio de unión al receptor (RBD) de la glicoproteína de pico (en adelante en inglés spike glycoprotein) (la spike glycoprotein es una proteína de membrana, en la membrana exterior del coronavirus, representada mediante triángulos azules sobre la superficie (tan familiar en nuestros días), que al sobresalir, da un aspecto de corona; de aquí el nombre de “coronavirus”); y (iii) al final del genoma, el propio gen NS8. Las secuencias de los coronavirus Bat SARS like (Bat-SL-CoV) (muestras ZXC21 y ZC45, números de acceso de la base de datos GenBank MG772934.1 y MG772933.1, respectivamente) fueron las únicas secuencias naturales de ADN que coincidieron perfectamente (match perfecto) con los marcadores de los genes Orf1ab y NS8. Por otro lado, el marcador del gen S no presentó ninguna coincidencia (match) con ninguna otra secuencia natural de ADN.
• Segundo, el análisis filogenético basado en las secuencias de genomas completos mostró que los coronavirus Bat-SL-CoV (ZXC21 y ZC45), Pangolin-CoV (muestra MP789, GenBank MT121216.1), BatCoV-RaTG13 (GenBank MN996532.1) y SARS -CoV-2 eran ortólogos. Es decir, se originaron a partir de un ancestro común, y se separaron entre sí mediante psoteriores eventos de especiación.
Es importante remarcar que no todos los coronavirus de pangolín se comportaron igual. Solo el coronavirus Panglolin-CoV (MP789) pertenece a este grupo de ortólogos. Otros coronavirus de pangolín estaban más distanciados filogenéticamente, consistentemente (soporte bootstrap 1000),
se agruparon en otra rama del árbol filogenético (Figura 1).
• Tercero, el análisis filogenético basado en secuencias de la spike glycoprotein (concretamente, desde el dominio N-terminal hasta el RBD, inclusive) mostró un patrón diferente. Las secuencias de BatCoV-RaTG13 y SARS-CoV-2 divergieron significativamente (soporte bootstrap 1000) de las secuencias de Bat-SL-CoV (ZXC21 y ZC45) y Pangolin-CoV (MP789). Además, los dos últimas se agruparon consistentemente (soporte bootstrap 1000) con otras secuencias de murciélagos. En este análisis filogenético, los otros coronavirus de pangolín también se agruparon consistentemente (soporte bootstrap 1000) en otra rama del árbol (cluster) (Figura 2).
¿Cómo y cuándo apareció el coronavirus de murciélago BatCoV-RaTG13?
El coronavirus BatCoV-RaTG13 se considera, de forma muy probable, que sea el progenitor directo del SARSCoV- 2 (1). Nuestros resultados también mostraron la estrecha relación filogenética entre ambas especies, sin embargo, ¿qué es lo que sabemos de este coronavirus de murciélago? El genoma de BatCoV-RaTG13 fue identificado y secuenciado por Zheng-Li Shi y colaboradores, 2020 (1), en el marco del estudio de un "nuevo coronavirus", que provocó la epidemia de síndrome respiratorio agudo en humanos en Wuhan, China (diciembre de 2019). Literalmente de (1): Descubrimos que una región corta de ARN polimerasa dependiente de ARN (RdRp) de un coronavirus de murciélago (BatCoV RaTG13), que se detectó previamente en Rhinolophus affinis de la provincia de Yunnan, mostró una alta identidad de secuencia con 2019-nCoV (SARSCoV-
2). Entonces, Zheng-Li Shi y colaboradores, 2020 (1), llevaron a cabo la secuenciación completa de los dos genomas de BatCoV-RaTG13 y SARS-CoV-2, encontrando una identidad de secuencia genómica entre ambos de 96,2%. La secuencia completa del genoma BatCoV-RaTG13 fue sometida e introducida a GenBank por Z.-L. Shi y colaboradores el 27 de enero de 2020. En la ficha de BatCoV-RaTG13 GenBank (MN996532.1),
aparece como fuente, muestra fecal; como huesped, Rhinolophus affinis; como país, China; y como fecha de recogida de la muestra, 24-jul-2013. Es decir, el coronavirus BatCoV RaTG13 fue aislado de una muestra fecal de murciélago en Yunnan en 2013, años antes de que se identificara por primera vez el SARS-CoV-2 y del inicio de la pandemia.
“Spike glycoprotein” de BatCoV-RaTG13: una incongruencia filogenética
En relación a la divergencia de BatCoV-RaTG13 y SARS-CoV-2 en la filogenia basada en las spike glicoproteins (Figura 1 y Figura 2), cabe mencionar que la región RBD se considera como la región más variable de los genomas del coronavirus (2). Sin embargo, como se muestra en el multialineamiento (Figura 3), la región del dominio N-terminal y del RBD está altamente conservada y muchos residuos de aminoácidos están
estrictamente conservados. De modo que, de acuerdo con la teoría de Motoo Kimura (3) sobre evolución molecular, en esta región tan crítica de los genomas del coronavirus, el equilibrio entre la deriva aleatoria y las constricciones funcionales debe haber estado muy comprometido, a lo largo de la evolución. No obstante, la sipke glicoproteína de SARS-CoV-2 se ha optimizado mucho para unirse al receptor humano ACE2 (2,4). ¡Por supuesto que sí! Es precisamente a través del RBD que el coronavirus se adhiere a la membrana celular e interactúa con el receptor del huésped, iniciando la infección (4,5).
No obstante, ¿qué evento evolutivo podría explicar esta extraordinaria optimización de la spike glycoprotein del SARS-CoV-2 para la infección humana, y también la incongruencia filogenética? Desde una perspectiva de evolución molecular, asumiendo que BatCoV-RaTG13 sea el progenitor directo del SARS-CoV-2, debemos situar este evento evolutivo en el genoma de BatCoV-RaTG13. Se podría considerar un evento de
recombinación entre BatCoV-RaTG13 y otros coronavirus, o una transferencia horizontal génica que reemplaza regiones variables en el gen S de BatCoV-RaTG13 (6,7). Pero, ya sea tanto en una recombinación o en una transferencia horizontal, siempre hay un “donador” del ADN que se recombina o se transfiere. En el presente estudio, no hemos podido identificar a ningún “donador” analizando la colección completa de nucleótidos del NCBI. Este razonamiento está de acuerdo con Zheng-Li Shi y colaboradores, 2020 (1), literalmente de (1): Usando multialineamientos de secuencias del genoma de 2019-nCoV, RaTG13, SARS-CoV y SARSr-CoV de murciélago reportados anteriormente, no se detectaron evidencias de eventos de recombinación en el genoma de 2019-nCoV (SARS-CoV-2).
El misterio del sitio de escisión de furina en la “spike glycoprotein” de SARS-CoV-2
En la secuencia de la spike glycoprotein de SARS-CoV-2 hay una pequeña inserción de cuatro aminoácidos, que no está en la de BatCoV-RaTG13. Esta pequeña inserción es responsable de la alta patogenicidad de la COVID-19. Esta inserción se conoce como “sitio polibásico de escisión de furina” (en adelante en inglés furin site). Es polibásico porque contiene aminoácidos básicos y es una pequeña región de la proteína que interacciona con la “furina”, otra proteína de membrana de las células humana (una proteasa), que favorece enormemente la infección del SARS-CoV-2.
Por otro lado, BatCoV-RaTG13 y SARS-CoV-2 también comparten otras tres pequeñas inserciones en el dominio N-terminal de la proteína (Figura 3). Dado que coronavirus de murciélago BatCoV-RaTG13 se aisló en 2013, es poco probable que tanto el ambos coronavirus hayan adquirido identicas inserciones en tres sitios distintos de la proteína (8). El hecho de compartir estas tres inserciones, apoya la hipótesis de que BatCoV-RaTG13 sea precursor de SARS-CoV-2. Se considera que el SARS-CoV-2 surgió directamente del murciélago al ser humano (1). Entonces, ¿cuándo ocurrió la inserción del furin site? Debe haber ocurrido en el huésped o durante la transmisión por recombinación, pero, no se han detectado eventos de recombinación en el SARS-CoV-2 (1).
Concretamente, el furin site, está constituido por los cuatro aminoácidos "PRRA", codificados por la inserción de 12 nucleótidos (CCT CGG CGG GCA) en la región conservada y bisagra del centro de escisión de las cadenas S1/S2 de la spike glycoprotein. A nivel de secuencia, el furin site se localiza 144 posiciones de aminoácidos aguas abajo del RBD (Figura 3 ). Por otra parte, el furin site tambiés se encuentra en otros betacoronavirus de otros linajes, pero no del linaje B, en el cual se ha clasificado el SARS-CoV-2 (9). A modo de ejemplo, a continuación se muestra un fragmento de un múltiple alineamiento de ecuencias de spike glycoprotein, de beta-coronavirus de otros linajes, que cubre la región del furin site (indicados en amarillo).
La presencia de un doblete de arginina es una característica estructural del sitio de unión a la furina (10):
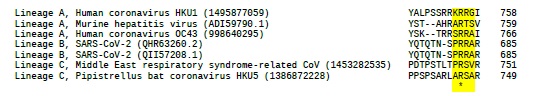 Como idea fundamental, el furin site es responsable de la alta infectividad y transmisibilidad de la COVID-19 (11). La interacción con la furina mejora la fusión célula-célula y media la fusión de la membrana. También está involucrada en otras enfermedades infecciosas y el cáncer (12), ahora, ee la clave de la COVID-19. Es a través de este sitio que el SARS-CoV-2 se ha optimizado realmente para unirse al receptor humano ACE2 e ingresar a las células humanas (13). El origen del furin site en el SARS-CoV-2 es desconcertante.
Como idea fundamental, el furin site es responsable de la alta infectividad y transmisibilidad de la COVID-19 (11). La interacción con la furina mejora la fusión célula-célula y media la fusión de la membrana. También está involucrada en otras enfermedades infecciosas y el cáncer (12), ahora, ee la clave de la COVID-19. Es a través de este sitio que el SARS-CoV-2 se ha optimizado realmente para unirse al receptor humano ACE2 e ingresar a las células humanas (13). El origen del furin site en el SARS-CoV-2 es desconcertante.
¿Existe algún animal huesped intermedio?
Inicialmente, se consideró que los pangolines eran el huésped intermedio (14). Sin embargo, en base a los presentes resultados y los descritos en la literatura, los análisis moleculares y filogenéticos no apoyan que el SARS-CoV-2 surgiera directamente de coronavirus de pangolín (15). Recientemente, C.M. Freuling et al., 2020 (16) también plantean la hipótesis de que los perros mapache (Nyctereutes procyonoides) podrían
haber sido huéspedes intermediarios del SARS-CoV-2, pero solo muestran que estos animales son susceptibles al coronavirus (16). Es un caso de zoonosis, donde probablemente el SARS-CoV-2 saltó entre humanos y animales no humanos. Otro caso de zoonosis ha sido el de Dinamarca que ha matado a 17 millones de visones para frenar una nueva variante del coronavirus que ha saltado a los humanos.
Entonces, a pesar de que el contacto directo entre humanos y murciélagos es limitado, y generalmente una especie intermedia a menudo está implicada en la transmisión de un virus emergentes de murciélagos a humanos (17), hasta la fecha, no es el caso en el SARS-CoV-2. O al menos, no se ha descubierto ningún huésped intermedio.
Interfaz entre biología y lógica
Los principios básicos de la biología también se cumplen en el mundo de los virus. De la Teoría Celular de Rudolf Virchow (1858), Omnis cellula ex cellula (cada célula deriva de otra célula preexistente), podría inferirse como "cada virus deriva de un virus preexistente". La selección natural de Charles Darwin (1859) sobre mutaciones y la lucha por la existencia es totalmente aplicable a los virus. El principio de Theodosius Dobzhansky (1973) "Nada en biología tiene sentido excepto a la luz de la evolución" (18) adquiere relevancia en el caso del origen del SARS-CoV-2. A partir de la información actual disponible en las bases de datos, es difícil encajar dicho origen en un modelo evolutivo racional.
La clasificación taxonómica del SARS2-CoV-2 también es confusa. Para abordar este punto, hemos considerado un criterio de genética forense, que se basa en una relación de probabilidades (likelihood ratio, LR), y tiene validez en un juicio. El LR es un parámetro que compara dos probabilidades de encontrar un mismo genotipo o marcadores genéticos de una evidencia, en dos personas distintas. El “LR” se utiliza
habitualmente para expresar los resultados de una prueba de ADN. Cuando un juez dicta sentencia sobre la culpabilidad de un sospechoso, basada en una prueba de ADN, requiere que la probabilidad de que el ADN de la evidencia sea del sospechoso, debe ser mucho mayor (billones superior, > 1012) que la probabilidad de que el mismo ADN sea de una persona aleatorio de la población a la que pertenece el sospechoso. Así, en genética forense, para que el LR sirvan de testigo de cargo, debe ser enormemente alto.
En el caso de la clasificación taxonómica del SARS2-CoV-2, hay una duda razonable de que pertenezca al grupo taxonómico, al cual se ha clasificado (linaje B del beta-coronavirus. Debido a la presencia del furin site,un LR es “infinito”. Esto es, la relación entre la probabilidad (P1) de que dada la secuencia actual del genoma del SARS-CoV-2, pertenezca a SARS-CoV-2, obviamente, es 1; y la probabilidad (P2) de que dada la misma secuencia del genoma del SARS-CoV-2, pertenezca a otro coronavirus de su grupo taxonómico, por el momento es 0 (porque el furin site solo se ha encontrado en el SARS-CoV-2). Entonces ese LR que compara las dos probabilidades P1 y P2 es “infinito” (LR = P1 / P2 = 1/0 = infinito). Esto es una forma probabilística de mostrar la dificultad de asociar el SARS-CoV-2 con el linaje B de los beta-coronavirus. En este sentido, Z.-L. Shi et al., 2020 (1), (sin tener en cuenta el furin site), también apuntaron esta duda. Literalmente de (1): El análisis filogenético del genoma completo y las secuencias de genes de RdRp y spike (S) mostró que, para todas las secuencias, RaTG13 es el pariente más cercano de 2019-nCoV (SARS-CoV-2) y forman un linaje distinto de otros SARSr-CoVs.
Dado que BatCoV-RaTG13 se considera el origen probable del SARS-CoV-2 (1), es sorprendente que después de casi un año de la pandemia de la COVID-19, no se hayan aislado más coronavirus de murciélago de la especie BatCoV-RaTG13. El murciélago es el reservorio natural de virus más estudiado. Por otro lado, los coronavirus de murciélago podrían aislarse de muestras fecales (¡no sería necesario un hisopo nasal de murciélago!). Una vez aislados, sus genomas completos se introducen en la base de datos GenBank.
Actualmente solo hay un genoma disponible de BatCoV-RaTG13 en GenBank (el referenciado, MN996532.1). Se requieren más genomas de BatCoV-RaTG13 para validar estadísticamente el 96,2% de identidad con el genoma de SARS-CoV-2. todavía queda un 3,8% de diferencia entre ambos, lo que puede ser clave para explicar la presencia del furin site en la spike glycoprotein del SARS-CoV-2. Por el contrario, actualmente hay
miles de genomas completos disponibles de SARS-CoV-2 en GenBank.
En la evolución biológica siempre hay "eslabones perdidos", en algunos casos, partir de descubrimientos de nuevos restos fósiles se pueden resolver. En el mundo de los virus no hay restos fósiles, pero los “eslabones perdidos” se pueden solucionar mediante el descubrimiento de nuevos virus. La presencia del furin site en el SARS-CoV-2 (en la parte más conservada de la proteína, y lejos de la región más variable) es señal de un "eslabón perdido" en nuestro conocimiento de su proceso evolutivo, y en definitiva de su origen. Este eslabón perdido también pone en duda que el BatCoV-RaTG13 sea el progenitor directo del SARS-CoV-2.
Como sucede en genética forense, que también se aplica en el mundo de los virus, cuando un marcador genético falla en una prueba de ADN, es suficiente para descartar una culpabilidad. Por lo tanto, existe una duda razonable sobre el origen del SARS-CoV-2, lo cual pode ser "un nuevo paradigma para la virología". La disponibilidad de nuevos genomas BatCoV-RaTG13 es esencial para poder abrir perspectivas. De lo contrario,
el origen del SARS-CoV-2 es un lado oscuro de la COVID-19. La duda puede hacer pensar que el SARS-CoV-2 sea un producto de laboratorio o un virus manipulado a propósito. La tecnología necesaria para ello está disponible. Para fines científicos y médicos, existen varios constructos sintéticos del genoma del SARS-CoV-2 en GenBank (MT108784.1, MT461669.1, MT461671.1, MT461670.1). Con fines terapéuticos, en 2008 se
creó un coronavirus recombinante sintético similar al SARS de murciélago, que resultó infeccioso en células cultivadas y en ratones (19). En este sentido, el debate debe continuar (20).
Como motivo de esperanza, por razones de probabilidad, y debido al mecanismo de "ensayo-error" de la evolución biológica, hay que pensar que un virus humano como el SARS-CoV-2, no volverá a surgir de la Madre Naturaleza por mucho, mucho tiempo (a nadie le toca el Gordo de la Lotería dos veces). Finalmente, hay que esperar una próxima vacuna y/o medicamentos efectivos para la COVID-19.
Antoni Romeu y Enric Ollé
Departamento de Bioquímica y Biotecnología, Universidad Rovira i Virgili, E-43007 Tarragona, España.
1: Catedrático de Bioquímica y Biología Molecular.
2: Profesor asociado.
Referencias
1. Peng Zhou, Xing-Lou Yang, Xian-Guang Wang, Ben Hu, Lei Zhang, Wei Zhang, Hao-Rui Si, Yan Zhu, Bei Li, Chao-Lin Huang, Hui-Dong Chen, Jing Chen, Yun Luo, Hua Guo, Ren-Di Jiang, Mei-Qin Liu, Ying Chen, Xu-Rui Shen, Xi Wang, Xiao-Shuang Zheng, Kai Zhao , Quan-Jiao Chen, Fei Deng, Lin-Lin Liu, Bing Yan, Fa-Xian Zhan, Yan-Yi Wang, Geng-Fu Xiao, Zheng-Li Shi. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579:270–273, 2020. PMID: 32015507. doi: 10.1038/s41586-020-2012-7.
2 . Kristian G Andersen, Andrew Rambaut, W Ian Lipkin, Edward C Holmes, Robert F Garry. The proximal origin of SARS-CoV-2. Nat. Med. 26:450-452, 2020. PMID: 32284615. doi: 10.1038/s41591-020-0820-9.
3. Kimura, M. The neutral theory of molecular evolution. Cambridge University, Cambridge. UK (1983).
4. Yushun Wan, Jian Shang, Rachel Graham, Ralph S Baric, Fang Li. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 94, e00127-20, 2020.PMID: 31996437. doi.org/10.1128/JVI.00127-20.
5. UNIPROT. P59594, Spike glycoprotein. Accessed October 09, 2020. https://www.uniprot.org/uniprot/P59594.
6 Dong-Sheng Chen, Yi-Quan Wu, Wei Zhang, San-Jie Jiang, Shan-Ze Chen. Horizontal gene transfer events reshape the global landscape of arm race between viruses and homo sapiens. Sci. Rep. 6:26934, 2016. PMID: 27270140. doi: 10.1038/srep26934.
7. Shahana S Malik, Syeda Azem-E-Zahra, Kyung Mo Kim, Gustavo Caetano-Anollés, Arshan Nasir. Do Viruses Exchange Genes across Superkingdoms of Life? Front. Microbiol. 8, 2110, 2017. PMID: 29163404. doi.org/10.3389/fmicb.2017.02110.
8. Xiaojun Li, Elena E Giorgi, Manukumar Honnayakanahalli Marichannegowda, Brian Foley, Chuan Xiao, Xiang-Peng Kong, Yue Chen, S Gnanakaran, Bette Korber, Feng Gao. Emergence of SARS-CoV-2 through recombination and strong purifying selection. Sci. Adv. 6(27):eabb9153. PMID: 32937441. doi: 10.1126/sciadv.abb9153.
9. Javier A Jaimes, Nicole M André, Joshua S Chappie, Jean K Millet, Gary R Whittaker. Phylogenetic Analysis and Structural Modeling of SARS-CoV-2 Spike Protein Reveals an Evolutionary Distinct and Proteolytically Sensitive Activation Loop. J. Mol. Biol. 432:3309-3325, 2020. PMID: 32320687. doi: 10.1016/j.jmb.2020.04.009.
10. Imène Kara, Marjorie Poggi, Bernadette Bonardo, Roland Govers, Jean-François Landrier, Sun Tian, Ingo Leibiger, Robert Day, John W M Creemers, Franck Peiretti. The Paired Basic Amino Acid-cleaving Enzyme 4 (PACE4) Is Involved in the Maturation of Insulin Receptor Isoform B. J. Biol. Chem. 290:2812–2821. PMID: 25527501. doi: 10.1074/jbc.M114.592543.
11. Shuai Xia, Qiaoshuai Lan, Shan Su, Xinling Wang, Wei Xu, Zezhong Liu, Yun Zhu, Qian Wang, Lu Lu, Shibo Jiang. The role of furin cleavage site in SARS-CoV-2 spike protein-mediated membrane fusion in the presence or absence of trypsin. Signal Transduct. Target Ther. 5:92, 2020. PMID: 32532959. doi.org/10.1038/s41392-020-0184-0.
12. Elisabeth Braun, Daniel Sauter. Furin-mediated protein processing in infectious diseases and cancer. Clin. Transl. Immunol. E1073, 2019. PMID: 31406574. doi.org/10.1002/cti2.1073.
13. Markus Hoffmann, Hannah Kleine-Weber, Stefan Pöhlmann. Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 78:779–784, 2020. PMID: 32362314. doi: 10.1016/j.molcel.2020.04.022.
14. Tao Zhang, Qunfu Wu, Zhigang Zhang. Probable Pangolin Origin of SARS-CoV-2 Associated with the COVID-19 Outbreak. Curr. Biol. 30:1346–1351.e2, 2020. PMID: 32315626. doi: 10.1016/j.cub.2020.03.022.
15. Ping Liu, Jing-Zhe Jiang, Xiu-Feng Wan, Yan Hua, Linmiao Li, Jiabin Zhou, Xiaohu Wang, Fanghui Hou, Jing
Chen, Jiejian Zou, Jinping Chen. Are pangolins the intermediate host of the 2019 novel coronavirus
(SARS-CoV-2)? PLoS Pathog. 16(5):e1008421, 2020. PMID: 32407364. doi:
10.1371/journal.ppat.1008421.
16. Conrad M. Freuling, Angele Breithaupt, Thomas Müller, Julia Sehl, Anne Balkema-Buschmann, Melanie Rissmann, Antonia Klein, Claudia Wylezich, Dirk Höper, Kerstin Wernike, Andrea Aebischer, Donata Hoffmann, Virginia Friedrichs, Anca Dorhoi, Martin H. Groschup, Martin Beer, Thomas C. Mettenleiter. Susceptibility of Raccoon Dogs for Experimental SARS-CoV-2 Infection. Emerg Infect Dis. 2020 Oct 22;26(12), 2020. PMID: 33089771. doi: 10.3201/eid2612.203733.
17. Shauna Milne-Price, Kerri L Miazgowicz, Vincent J Munster. The emergence of the Middle East Respiratory Syndrome coronavirus. Pathog. Dis. 71:21-176, 2014. PMID: 24585737. doi: 10.1111/2049- 632X.12166.
18. Theodosius Dobzhansky. Nothing in Biology Makes Sense except in the Light of Evolution. The American Biology Teacher 35:125-129, 1973.
19. Michelle M Becker , Rachel L Graham, Eric F Donaldson, Barry Rockx, Amy C Sims, Timothy Sheahan, Raymond J Pickles, Davide Corti, Robert E Johnston, Ralph S Baric, Mark R Denison. Synthetic recombinant bat SARS-like coronavirus is infectious in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 105:19944–19949, 2008. PMID: 19036930. doi: 10.1073/pnas.0808116105.
20. Heidi J Larson. A lack of information can become misinformation. Nature 580:306, 2020. PMID: 32231320. doi: 10.1038/d41586-020-00920-w.
21. Muhamad Fahmi, Yukihiko Kubota, Masahiro Ito. Nonstructural proteins NS7b and NS8 are likely to be phylogenetically associated with evolution of 2019-nCoV. Infect. Genet. Evol. 81:104272, 2020. PMID: 32142938. doi.org/10.1016/j.meegid.2020.104272.
22. Fábio Madeira, Young Mi Park, Joon Lee, Nicola Buso, Tamer Gur, Nandana Madhusoodanan, Prasad Basutkar, Adrian R N Tivey, Simon C Potter, Robert D Finn, Rodrigo Lopez. The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucl. Acids Res. 47(W1):W636-W641, 2019. PMID: 30976793. doi: 10.1093/nar/gkz268.
23. Ivica Letunic, Peer Bork. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucl. Acids Res. 2011, Vol. 39(W475–W478), 2011. PMID: 21470960. doi:10.1093/nar/gkr201.
24. Jun Lan, Jiwan Ge, Jinfang Yu, Sisi Shan, Huan Zhou, Shilong Fan, Qi Zhang, Xuanling Shi, Qisheng Wang, Linqi Zhang, Xinquan Wang. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 581:215-220, 2020. PMID: 32225176. doi:10.1038/s41586-020-2180-5.
Agradecimientos
Este trabajo no ha recibido subvenciones de ninguna institución de apoyo a la investigación.
Declaración de conflicto de intereses
Los autores declaran no tener ningún conflicto de intereses.
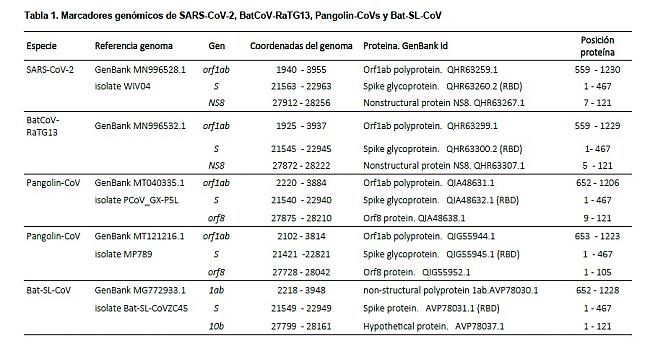
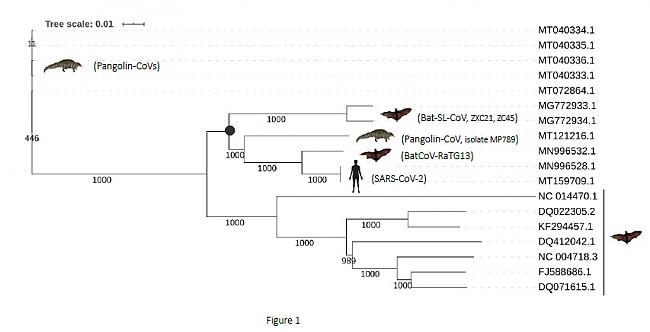
Figura 1. Árbol filogenético de una muestra de coronavirus basado en secuencias de genomas completos
Árbol filogenético basado en un alineamiento múltiple de una selección de genomas completos de coronavirus. La muestra incluye el BatCoV-RaTG13, los coronavirus de pangolín disponibles, una selección de coronavirus de murciélago extraídos de la literatura (1,14,21) y dos genomas del SARS-CoV-2 como representantes del grupo taxonómico: NCBI, Severe acute respiratory syndrome coronavirus 2 (taxid:2697049). El árbol filogenético se construyó utilizando el método Neighbor Joining del software Clustal Omega (v.1.2.4) con parámetros por defecto (22) y la herramienta iTol Interactive Tree Of Life (23). La consistencia de las agrupaciones se calculó mediante bootstrap utilizando 1000 repeticiones. La barra de escala de árbol representa la distancia evolutiva. El punto negro indica el ancestro común del grupo de coronavirus relacionados con la COVID-19. El número de acceso de GenBank de los genomas completos y el coronavirus son los siguientes (en la misma disposición que en el árbol filogenético): MT040333.1 a MT040336.1, coronavirus pangolín; MG772933.1 y MG772934.1, coronavirus de tipo murciélago SARS; MT121216.1 (aislado MP789) Coronavirus de pangolín; MN996532.1, BatCoV-RaTG13; MN996528.1 y MT159709.1, SARS-CoV-2; NC_014470.1, Coronavirus de murciélago BM48-31/BGR/2008; DQ022305.2, coronavirus HKU3-1 del SARS de murciélago; KF294457.1, coronavirus de tipo murciélago SARS; DQ412042.1, Bat SARS CoV Rf1/2004; NC_004718.3, coronavirus Tor2 del SARS; FJ588686.1, coronavirus del SARS Rs_672/2006; DQ071615.1, Bat SARS CoV Rp3/2004.
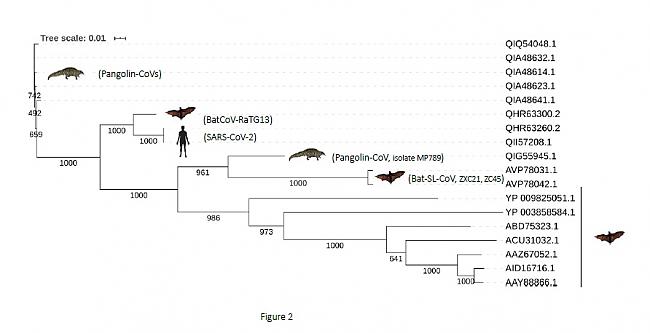
Figura 2. Árbol filogenético de una muestra coronavirus basado en secuencias de spike glycoprotein
Árbol filogenético basado en el multialineamiento de la región más variable de la spike glycoprotein (desde el dominio N-terminal hasta el RBD). La muestra incluye las secuencias de los mismos coronavirus de la Figura 1, con el objetivo de comparar el comportamiento filogenético, en función del tipo de seciencia. La posición de RBD se basó en (24). El árbol filogenético se construyó utilizando el método Neighbor Joining del software
Clustal Omega (v.1.2.4), con parámetros por defecto (22) y la herramienta iTol Interactive Tree Of Life (23).
La consistencia de las agrupaciones se calculó mediante bootstrap utilizando 1000 repeticiones. La barra de escala de árbol representa la distancia evolutiva. El número de acceso de GenBank de la spike glycoprotein y el correspondiente coronavirus con los siguientes (en la misma disposición que en el árbol filogenético): QIA48641.1, QIA48632.1, QIA48614.1, QIA48623.1, QIQ54048.1, coronavirus pangolín; QHR63300.2,
coronavirus de murciélago RaTG13; QHR63260.2, QII57208.1, SARS-CoV-2; QIG55945.1, coronavirus de pangolín; AVP78031.1, AVP78042.1, coronavirus de tipo murciélago SARS; YP_009825051.1, coronavirus Tor2 del SARS; YP_003858584.1, Coronavirus de murciélago BM48-31/BGR/2008; ABD75323.1, Bat SARS CoV Rf1/2004; ACU31032.1, coronavirus del SARS Rs_672/2006; AAZ67052.1, Bat SARS CoV Rp3/2004;
AID16716.1, coronavirus de tipo murciélago SARS; AAY88866.1, coronavirus HKU3-1 del SARS de murciélago. 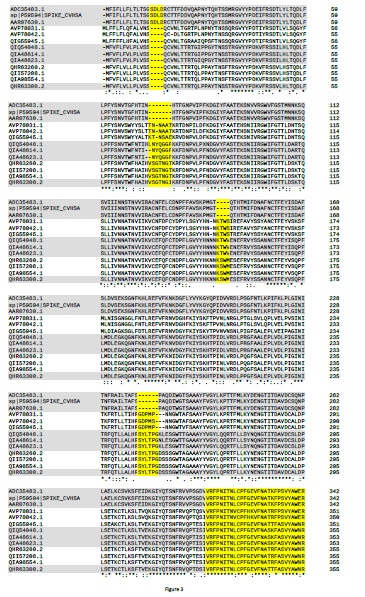
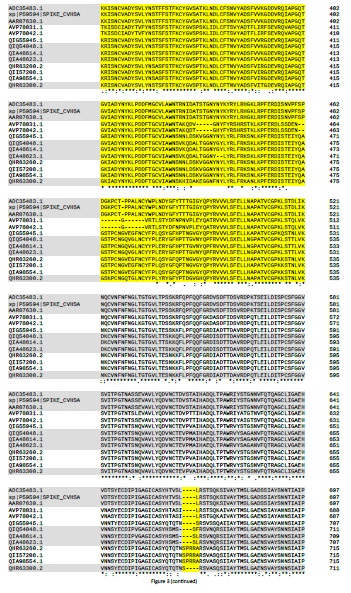
Figura 3. Multialneamiento de spike glycoprotein de una selección de coronavirus.
Alineamiento múltiple de secuencia spike glycoprotein. Los grupos de las muestras seleccionadas son: (i) coronavirus del SARS humano (SARS-CoV) (longitud de 1255 amoniacidos); (ii) Bat-SL-CoV (1245); (iii) Pangolin-CoVs (1265-1269); (iv) SARS-CoV-2 (1273); y (v) BatCoV-RaTG13 (1269). Para visualizar mejor las características de cada grupo, solo hay tres secuencias representativas de cada uno de ellos. El multialineamiento se construyó mediante el software Clustal Omega (v.1.2.4), con los parámetros por defecto (22). Los aminoácidos estrictamente conservados se indican con *, los espacios con -. La posición de los aminoácidos en cada secuencia está indicada por los números a la derecha. Los espacios correspondientes a la delección y las tres inserciones características de las secuencias relacionadas con COVID-19, el RBD y el sitio de escisión de furina (furin site) del SARS-CoV-2 están resaltados en amarillo. La banda de colores (gris-blanco) tiene por objeto resaltar las características de secuencia de: SARS-CoV; Bat-SLCoV y Pangolin-CoV (MP789); otros Pangolin-CoV; SARS-CoV-2; y BatCoV-RaTG13. La posición de RBD se basó en (24). La figura solo se muestra hasta el furin site. Hasta el extremo C-terminal de la proteína, la mayoría de las posiciones eran estrictamente conservadas. El número de acceso de GenBank de las secuencias de spike glycoprotein y los respectivos coronavirus son los siguientes: ADC35483.1, coronavirus del SARS HKU-39849; sp | P59594 | (UNIPROT SPIKE_CVHSA); AAR07630.1, coronavirus BJ302 del SARS; AVP78031.1, Bat-SL-CoV ZC45; AVP78042.1, Bat-SL-CoV ZXC21; QIG55945.1, PangolinCoV, MP789; QIQ54048.1, Pang-CoV, GX-P2V; QIA48614.1, Pang-CoV, GX-P4L; QIA48623.1, Pang-CoV, GX-P1E; QHR63260.2, SARS-CoV-2; QII57208.1 SARSCoV-
2; QIA98554.1, SARS-CoV-2; QHR63300.2, BatCoV-RaTG13.